Python API¶
In summary, Hydrides provides two functionalities:
Adding hydrogen atoms to a structure with missing hydrogen atoms
Relaxing hydrogen positions in a structure with existing hydrogen atoms
Typically both functionalities are combined: In a first step hydrogen atoms are added to a molecular model and then the hydrogen atoms are relaxed in the resulting model.
Note that there are legit cases for using only one of these functionalities: The initial placement of the hydrogen atoms might be sufficient for your use case, so no relaxation of the rotatable hydrogen atoms is required. Or you might have an existing structure containing hydrogen atoms and you want to reduce atom clashes and reconstruct hydrogen bonds.
To illustrate the effects of each step, this tutorial will add hydrogen atoms
to a molecular model of 2-nitrophenol, where hydrogen atoms are missing.
Hydride expands on data types from
Biotite, so an AtomArray is used
to represent a molecular model, including it atom descriptions, coordinates and
bonds.
If you are new to Biotite, have a look at the
official documentation.
For this example the structure of 2-nitrophenol is loaded from a MOL file.
import biotite.structure.io.mol as mol
mol_file = mol.MOLFile.read("path/to/nitrophenol.mol")
molecule = mol_file.get_structure()
print(type(molecule))
print()
print(molecule)
print(molecule.bonds.as_array())
<class 'biotite.structure.AtomArray'>
0 O 2.244 0.777 0.012
0 N 1.635 -0.278 0.006
0 O 2.245 -1.333 -0.004
0 C 0.156 -0.278 0.006
0 C -0.539 0.923 0.017
0 O 0.138 2.100 0.029
0 C -0.535 -1.474 -0.012
0 C -1.919 -1.475 -0.013
0 C -2.613 -0.280 0.003
0 C -1.926 0.919 0.016
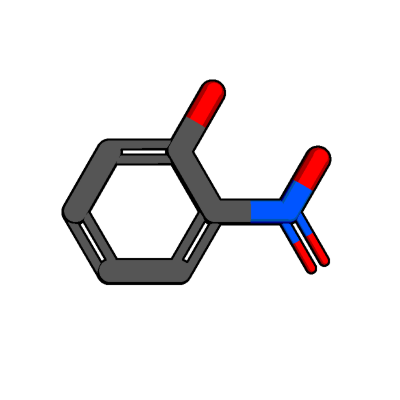
As you can see, this molecule does not contain any hydrogen atoms, yet.
This circumstance is changed by add_hydrogen().
Note that the input AtomArray must not already contain hydrogen atoms.
If at some places hydrogen atoms already exist, these must be removed.
Furthermore, the AtomArray must have an associated charge
attribute, containing the formal charges, and an associated BondList.
import hydride
# Remove already present hydrogen atoms (only necessary in rare cases)
molecule = molecule[molecule.element != "H"]
# Add hydrogen atoms
molecule, mask = hydride.add_hydrogen(molecule)
print(molecule)
print()
print(mask)
0 O 2.244 0.777 0.012
0 N 1.635 -0.278 0.006
0 O 2.245 -1.333 -0.004
0 C 0.156 -0.278 0.006
0 C -0.539 0.923 0.017
0 O 0.138 2.100 0.029
0 C -0.535 -1.474 -0.012
0 C -1.919 -1.475 -0.013
0 C -2.613 -0.280 0.003
0 C -1.926 0.919 0.016
0 H -0.180 2.650 0.766
0 H -0.034 -2.435 -0.026
0 H -2.419 -2.437 -0.026
0 H -3.696 -0.236 0.006
0 H -2.511 1.832 0.025
[ True True True True True True True True True True False False
False False False]
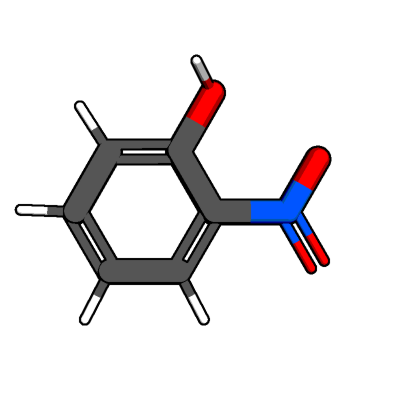
add_hydrogen() returns two objects:
The AtomArray, including the input atoms plus the added hydrogen
atoms, and a boolean mask (a numpy.ndarray), that selects the original
input atoms.
This means, if the mask is applied to the returned AtomArray, the
hydrogen atoms are removed again.
print(molecule[mask])
0 O 2.244 0.777 0.012
0 N 1.635 -0.278 0.006
0 O 2.245 -1.333 -0.004
0 C 0.156 -0.278 0.006
0 C -0.539 0.923 0.017
0 O 0.138 2.100 0.029
0 C -0.535 -1.474 -0.012
0 C -1.919 -1.475 -0.013
0 C -2.613 -0.280 0.003
0 C -1.926 0.919 0.016
Regarding the order of atoms in the returned AtomArray, the hydrogen
atoms are placed behind the heavy atoms for each residue/molecule separately.
Most hydrogen atoms are already placed correctly with respect to bond angles
and lengths.
However, the hydrogen atom in the hydroxy group is not in a energy-minimized
state, as an intramolecular hydrogen bond to the nitro group would be expected.
The energy minimization is performed with relax_hydrogen().
molecule.coord = hydride.relax_hydrogen(molecule)
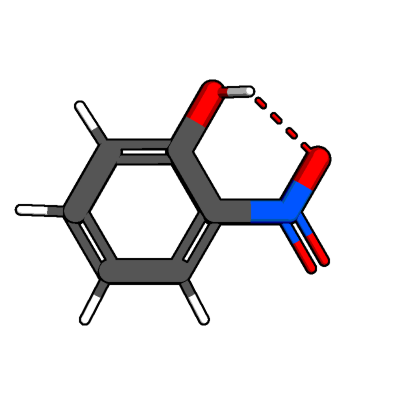
relax_hydrogen() is able to optimize the position of hydrogen atoms at
terminal groups.
In this case it was able to orient the hydrogen atom at the hydroxy group
towards the nitro group to form a hydrogen bond.
Custom fragment and atom name libraries¶
add_hydrogen() uses a FragmentLibrary to find the correct
number and positions of hydrogen atoms for each heavy atom in the input
AtomArray and a AtomNameLibrary to find
the correct atom name (e.g. 'HA').
The default fragment library (FragmentLibrary.standard_library())
contains all fragments from the entire
Chemical Component Dictionary and the
default atom name library (AtomNameLibrary.standard_library())
contains atom names for all amino acids and standard nucleotides.
Furthermore, the atom name library provides a reasonable naming scheme for
unknown residues/molecules.
Therefore, it is seldom required to provide a custom library.
However, in rare cases the fragment library might not contain a required
fragment or the automatic atom naming is not sufficient.
A custom FragmentLibrary can be created either via its constructor
or by copying the default library.
library = copy.deepcopy(hydride.FragmentLibrary.standard_library())
Then molecular models containing hydrogen atoms are added to the library. Internally, a model is split into fragments and these fragments are stored in the library’s internal dictionary. Hence, such a molecular model act as template for the later hydrogen addition.
library.add_molecule(template_molecule)
Finally the fragment library can be provided to add_hydrogen().
hydride.add_hydrogen(molecule, fragment_library=library)
In a similar way, a custom AtomNameLibrary can be created, filled with
template molecules and used in add_hydrogen().
library = copy.deepcopy(hydride.AtomNameLibrary.standard_library())
library.add_molecule(template_molecule)
hydride.add_hydrogen(molecule, name_library=library)
Handling periodic boundary conditions¶
By default, add_hydrogen() and relax_hydrogen() do not take
periodic boundary conditions into account, as they appear e.g. in MD
simulations.
Consequently, interactions over the periodic boundary are not taken into
account and, more importantly, hydrogen atoms are not placed correctly, if the
molecule is divided by the boundary.
To tell Hydride to consider periodic boundary conditions the box parameter
needs to be provided.
The value can be either a an array of the three box vectors or True, in
which case the box is taken from the input structure.
molecule, _ = hydride.add_hydrogen(molecule, box=True)
molecule.coord = hydride.relax_hydrogen(molecule, box=True)
Note that this slows down the addition and relaxation procedure.
Tweaking relaxation speed and accuracy¶
Usually, the relaxation is fast and accurate enough for most applications. However, the user is able to adjust some parameters to shift the speed-accuracy-balance to either side.
By default, relax_hydrogen() runs until the energy of the conformation
does not improve anymore.
However, the maximum number of iterations can also be given with the
iterations parameter.
If the number of relaxation steps reaches this value, the relaxation terminates
regardless of whether an energy minimum is attained.
The angle_increment parameter controls the angle by which each rotatable
bond is changed in each relaxation iteration. Lowering this value leads to a
higher resolution of the returned conformation, but more iterations are
required to find the optimum.
Observing the relaxation¶
In order to get insights into the course of the relaxation, the user can
optionally obtain the coordinates and energy values for each iteration via the
return_trajectory and return_energies parameters, respectively.
import matplotlib.pyplot as plt
molecule_with_h, mask = hydride.add_hydrogen(molecule)
coord, energies = hydride.relax_hydrogen(
molecule_with_h, return_trajectory=True, return_energies=True
)
print(coord.shape)
print(energies.shape)
fig, ax = plt.subplots(figsize=(4.0, 2.0))
ax.plot(energies)
ax.set_xlabel("Iteration")
ax.set_ylabel("Energy")
fig.tight_layout()
Handling missing formal charges¶
Both add_hydrogen() and relax_hydrogen() require associated
formal charges for correct fragment identification or the electrostatic
potential, respectively.
However, for many entries in the RCSB PDB they are not properly set.
At least for canonical amino acids this issue can be remedied with
estimate_amino_acid_charges().
This function calculates the formal charges of atoms in amino acids based on a
given pH value.
charges = hydride.estimate_amino_acid_charges(molecule, ph=7.0)
molecule.set_annotation("charge", charges)
Handling missing bonds¶
The input AtomArray is also required to have an associated
BondList.
Unfortunately, this information cannot be retrieved from PDB or PDBX/mmCIF
files.
If the residues in the input structure are part of the
Chemical Component Dictionary, the BondList can be created with
biotite.structure.connect_via_residue_names().
import biotite.structure as struc
molecule.bonds = struc.connect_via_residue_names(molecule)
Classes and functions¶
- class hydride.FragmentLibrary¶
A molecule fragment library for estimation of hydrogen positions.
For each molecule added to the
FragmentLibrary, the molecule is split into fragments. Each fragment consists ofA central heavy atom,
bond order and position of its bonded heavy atoms and
and positions of bonded hydrogen atoms.
The properties of the fragment (central atom element, central atom charge, order of connected bonds) are stored in a dictionary mapping these properties to heavy and hydrogen atom positions.
If hydrogen atoms should be added to a target structure, the target structure is also split into fragments. Now the corresponding reference fragment in the library dictionary is accessed for each fragment. The corresponding atom coordinates of the reference fragment are superimposed [1] [2] onto the target fragment to obtain the hydrogen coordinates for the heavy atom.
The constructor of this class creates an empty library.
References
[1]W Kabsch, “A solution for the best rotation to relate two sets of vectors.” Acta Cryst, 32, 922-923 (1976).
[2]W Kabsch, “A discussion of the solution for the best rotation to relate two sets of vectors.” Acta Cryst, 34, 827-828 (1978).
- add_molecule(molecule)¶
Add the fragments of a molecule to the library.
- Parameters:
- moleculeAtomArray
A molecule, whose fragments should be added to the library. The structure must contain hydrogen atoms for each applicable heavy atom. The molecule must have an associated
BondList. The molecule must also include the charge annotation array, depicting the formal charge for each atom.
- calculate_hydrogen_coord(atoms, mask=None, box=None)¶
Estimate the hydrogen coordinates for each atom in a given structure/molecule.
- Parameters:
- atomsAtomArray, shape=(n,)
The structure to get the hydrogen atom positions for. The structure must not contain any hydrogen atoms. The structure must have an associated
BondList. The structure must also include the charge annotation array, depicting the formal charge for each atom.- maskndarray, shape=(n,), dtype=bool
A boolean mask that is true for each heavy atom, where corresponsing hydrogen atom positions should be calculated. By default, hydrogen atoms are calculated for all applicable atoms.
- boxbool or array-like, shape=(3,3), dtype=float, optional
If this parameter is set, periodic boundary conditions are taken into account (minimum-image convention), based on the box vectors given with this parameter. If box is set to true, the box vectors are taken from the
boxattribute of atoms instead.
- Returns:
- hydrogen_coordlist of (ndarray, shape=(k,3), dtype=np.float32), length=n
A list of hydrogen coordinates for each atom in the input atoms. k is the number of hydrogen atoms for this atom. Each atom, that is not included in the input mask or which has no fitting fragment in the library, has no corresponding hydrogen coordinates, i.e. k is 0.
- static standard_library()¶
Get the standard
FragmentLibrary. The library contains fragments from all molecules in the RCSB Chemical Component Dictionary.- Returns:
- libraryFragmentLibrary
The standard library.
- class hydride.AtomNameLibrary¶
A library for generating hydrogen atom names.
For each molecule added to the
AtomNameLibrary, the hydrogen atom names are saved for each heavy atom in this molecule.If hydrogen atom names should be generated for a heavy atom, the library first looks for a corresponding entry in the library. If such entry is not found, since the molecule was never added to the library, the hydrogen atom names are guessed based on common hydrogen naming schemes.
- add_molecule(molecule)¶
Add the hydrogen atom names for each heavy atom in the molecule to the library.
- Parameters:
- moleculeAtomArray
The molecule to use the hydrogen atom names from.
- generate_hydrogen_names(heavy_res_name, heavy_atom_name)¶
Generate hydrogen atom names for the given residue and heavy atom name.
If the residue is not found in the library, the hydrogen atom name is guessed based on common hydrogen naming schemes.
- static standard_library()¶
Get the standard
AtomNameLibrary. The library contains atom names for the most prominent molecules including amino acids and nucleotides.- Returns:
- libraryAtomNameLibrary
The standard library.
- hydride.add_hydrogen(atoms, mask=None, fragment_library=None, name_library=None, box=None)¶
Add hydrogen atoms to a structure.
The hydrogen atoms for each residue are placed directly behind the atoms from this residue. The function also tries to assign the correct atom name to each added hydrogen atom.
- Parameters:
- atomsAtomArray, shape=(n,)
The structure, where hydrogen atoms should be added. The structure must have an associated
BondList. The structure must also include the charge annotation array, depicting the formal charge for each atom. The structure must not have any annotated hydrogen atoms (in the region covered by mask), yet.- maskndarray, shape=(n,), dtype=bool, optional
A boolean mask that is true for each atom, where hydrogen atoms should be added. By default, hydrogen atoms are added to all applicable atoms.
- fragment_libraryFragmentLibrary
The fragment library to use for hydrogen position estimation. By default
FragmentLibrary.standard_library()is used, containing fragments from all molecules in the RCSB Chemical Component Dictionary.- name_libraryAtomNameLibrary
The atom name library to use for hydrogen naming. By default
AtomNameLibrary.standard_library()is used, containing atom names for the most prominent molecules including amino acids and nucleotides. For all other molecules the hydrogen atom names are guessed.- boxbool or array-like, shape=(3,3), dtype=float, optional
If this parameter is set, periodic boundary conditions are taken into account (minimum-image convention), based on the box vectors given with this parameter. If box is set to true, the box vectors are taken from the
boxattribute of atoms instead.
- Returns:
- hydrogenated_atomsAtomArray, shape=(p,)
The atoms from the input atoms with additional hydrogen atoms. Although, the hydrogen positions are meaningful with respect to bond lengths and angles, the dihedral angles are not optimized.
- original_atom_maskndarray, shape=(p,)
A boolean mask that is true for each atom in hydrogenated_atoms that originates from the input atoms. That means, that if the mask is applied to hydrogenated_atoms, the input structure is restored.
- hydride.relax_hydrogen(atoms, iterations=None, angle_increment=np.deg2rad(10), return_trajectory=False, return_energies=False, partial_charges=None, box=None)¶
Optimize the hydrogen atom positions by rotating about terminal bonds. The relaxation uses hill climbing based on an electrostatic and a nonbonded potential [1].
- Parameters:
- atomsAtomArray, shape=(n,)
The structure, whose hydrogen atoms should be relaxed. Must have an associated
BondList. Note thatBondType.ANYbonds are not considered for rotation- iterationsint, optional
Limit the number of relaxation iterations. The runtime scales approximately linearly with the number of iterations. By default, the relaxation runs until a local optimum has been found, i.e. the hydrogen coordinates do not change anymore. If this parameter is set, the relaxation terminates before this point after the given number of interations.
- maskndarray, shape=(n,), dtype=bool
Ignore bonds, where the index of the heavy atom in the mask is False. By default no bonds are ignored.
- angle_incrementfloat, optional
The angle in radians by which a bond can be rotated in each iteration. Smaller values increase the accurucy, but increase the number of required iterations.
- return_trajectorybool, optional
If set to true, the resulting coordinates for each relaxation step are returned, instead of the coordinates of the final step.
- return_energiesbool, optional
If set to true, also the calculated energy for the conformation of each relaxation step is returned. This parameter can be useful for monitoring and debugging.
- partial_chargesndarray, shape=(n,), dtype=float, optional
The partial charges for each atom used to calculate Coulomb interactions. By default the charges are calculated using
biotite.structure.partial_charges().- boxbool or array-like, shape=(3,3), dtype=float, optional
If this parameter is set, periodic boundary conditions are taken into account (minimum-image convention), based on the box vectors given with this parameter. If box is set to true, the box vectors are taken from the
boxattribute of atoms instead.
- Returns:
- relaxed_coordndarray, shape=(n,) or shape=(m,n), dtype=np.float32
The optimized coordinates. The coordinates for all heavy atoms remain unchanged. if return_trajectory is set to true, not only the coordinates after relaxation, but the coordinates from each step are returned.
- energiesndarray, shape=(m,), dtype=np.float32
The energy for each step. Only returned, if return_energies is set to true
Notes
The potential consists of the following terms:
\[ \begin{align}\begin{aligned}V = V_\text{el} + V_\text{nb}\\V_\text{el} = 332.067 \sum_i^\text{H} \sum_j^\text{All} \frac{q_i q_j}{D_{ij}}\\E_\text{nb} = \epsilon_{ij} \sum_i^\text{H} \sum_j^\text{All} \left( \frac{r_{ij}^{12}}{D_{ij}^{12}} - 2\frac{r_{ij}^6}{D_{ij}^6} \right)\end{aligned}\end{align} \]where \(D_{ij}\) is the distance between the atoms \(i\) and \(j\). \(\epsilon_{ij}\) and \(r_{ij}\) are the well depth and optimal distance between these atoms, respectively, and are calculated as
\[ \begin{align}\begin{aligned}\epsilon_{ij} = \sqrt{ \epsilon_i \epsilon_j},\\r_{ij} = \frac{r_i + r_j}{2}.\end{aligned}\end{align} \]\(\epsilon_{i/j}\) and \(r_{i/j}\) are taken from the Universal Force Field [1] [2].
References
[1] (1,2)AK Rappé, CJ Casewit, KS Colwell, WA Goddard III and WM Skiff, “UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations.” J Am Chem Soc, 114, 10024-10035 (1992).
[2]T Ogawa and T Nakano, “The Extended Universal Force Field (XUFF): Theory and applications.” CBIJ, 10, 111-133 (2010)
- hydride.estimate_amino_acid_charges(atoms, ph)¶
Estimate the charge of heavy atoms in peptides based on the protonation state of the free amino acid [1].
- Parameters:
- atomsAtomArray, shape=(n,) or AtomArrayStack, shape=(m,n)
The atoms to calculate the charges for.
- phfloat
The charges are estimated based on this pH value.
- Returns:
- chargesndarray, shape=(n,), dtype=int
The estimated charges. 0 for all atoms that are not part of an amino acid.
Notes
The protonation state is only estimated for canonical amino acids.
References
[1]DR Lide, “CRC Handbook of Chemistry and Physics.” CRC Press, (2003).
